

This page includes selected results from our research. You can navigate between different subjects listed below. For a complete list of publications go to the publications page.
Infrared Phosphorene

Black phosphorus is an infrared layered material. Its bandgap complements other widely studied two-dimensional materials: zero-gap graphene and visible/near-infrared gap transition metal dichalcogenides. Although highly desirable, a comprehensive infrared characterization is still lacking. Here we report a systematic infrared study of mechanically exfoliated few-layer black phosphorus, with thickness ranging from 2 to 15 layers and photon energy spanning from 0.25 to 1.36 eV. Each few-layer black phosphorus exhibits a thickness-dependent unique infrared spectrum with a series of absorption resonances, which reveals the underlying electronic structure evolution and serves as its infrared fingerprints. Surprisingly, unexpected absorption features, which are associated with the forbidden optical transitions, have been observed. Furthermore, we unambiguously demonstrate that controllable uniaxial strain can be used as a convenient and effective approach to tune the electronic structure of few-layer black phosphorus. Our study paves the way for black phosphorus applications in infrared photonics and optoelectronics.
Environmental Stability of Tellurium-Containing Nanomaterials

Recent studies have shown that tellurium-based two-dimensional (2D) crystals undergo dramatic structural, physical, and chemical changes under ambient conditions, which adversely impact their much desired properties. Here, we introduce a diazonium molecule functionalization-based surface engineering route that greatly enhances their environmental stability without sacrificing their much desired properties. Spectroscopy and microscopy results show that diazonium groups significantly slow down the surface reactions, and consequently, gallium telluride (GaTe), zirconium telluride (ZrTe3), and molybdenum ditelluride (MoTe2) gain strong resistance to surface transformation in air or when immersed under water. Density functional theory calculations show that functionalizing molecules reduce surface reactivity of Te-containing 2D surfaces by chemical binding followed by an electron withdrawal process. While pristine surfaces structurally decompose because of strong reactivity of Te surface atoms, passivated functionalized surfaces retain their structural anisotropy, optical band gap, and emission characteristics as evidenced by our conductive atomic force microscopy, photoluminescence, and absorption spectroscopy measurements. Overall, our findings offer an effective method to increase the stability of these environmentally sensitive materials without impacting much of their physical properties.
Self Healing of Defects in 2D Materials
Self-healing mechanisms of vacancy defects in graphene and silicene are studied using first-principles calculations. We investigated host adatom adsorption, diffusion, vacancy formation, and revealed atomistic mechanisms in the healing of single, double, and triple vacancies of single-layer graphene and silicene. Silicon adatom, which is adsorbed to silicene at the top site forms a dumbbell-like structure by pushing one Si atom underneath. The asymmetric reconstruction of the single vacancy in graphene is induced by the magnetization through the rebonding of two dangling bonds and acquiring a significant magnetic moment through the remaining unsaturated dangling bond. In silicene, three twofold coordinated atoms surrounding the single vacancy become fourfold coordinated and nonmagnetic through rebonding. The energy gained through new bond formation becomes the driving force for the reconstruction. Under the external supply of host atoms, while the vacancy defects of graphene heal perfectly, the Stone-Wales defect can form in the course of healing of silicene vacancy. The electronic and magnetic properties of suspended, single-layer graphene and silicene are modified by reconstructed vacancy defects.

Dissociation of Molecules at Defect Sites
We study the interaction of molecules with the vacancy defects of graphene and silicene. Atoms around the bare vacancy reconstruct and specific chemically active sites are created. Although H2, O2 and CO remain intact on both pristine graphene and silicene, these molecules can dissociate when they are placed at the close proximity of these chemically active sites and nucleate centers for the hydrogenation and oxygenation. Saturation of the dangling bonds at the defect sites by constituent atoms of dissociated molecules gives rise to significant modification of electronic and magnetic properties. We analyzed the mechanism of the dissociation and revealed a concerted action of surrounding host atoms together with dissociated molecules to lower the energy barrier needed for dissociation. The dissociations of H2O and OH are hindered by high energy barriers. Our study suggests that graphene and silicene can be functionalized by creating meshes of single vacancy, where specific molecules can dissociate, and some other molecules can be pinned.

Superlubricity
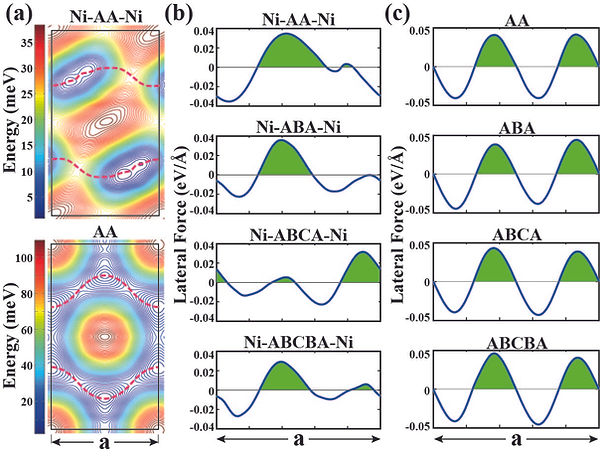
A single graphene layer placed between two parallel Ni(111) surfaces screens the strong attractive force and results in a significant reduction of adhesion and sliding friction. When two graphene layers are inserted, each graphene is attached to one of the metal surfaces with a significant binding and reduces the adhesion further. In the sliding motion of these surfaces the transition from stick-slip to continuous sliding is attained, whereby nonequilibrium phonon generation through sudden processes is suppressed. The adhesion and corrugation strength continues to decrease upon insertion of the third graphene layer and eventually saturates at a constant value with increasing number of graphene layers. In the absence of Ni surfaces, the corrugation strength of multilayered graphene is relatively higher and practically independent of the number of layers. Present first-principles calculations reveal the superlubricant feature of graphene layers placed between pseudomorphic Ni(111) surfaces, which is achieved through the coupling of Ni-3d and graphene-π orbitals. The effect of graphene layers inserted between a pair of parallel Cu(111) and Al(111) surfaces is also discussed. The treatment of sliding friction under the constant loading force, by taking into account the deformations corresponding to any relative positions of sliding slabs, is the unique feature of our study.
Atomic Chains

Nucleation and growth mechanisms of short chains of carbon atoms on single-layer, hexagonal boron nitride (h-BN) and short BN chains on graphene are investigated using first-principles plane-wave calculations. Our analysis starts with the adsorption of a single carbon adatom and examines its migrations. Once a C2 nucleates on h-BN, the insertion of each additional carbon at its close proximity causes a short segment of carbon atomic chain to grow by one atom at at a time in a quaint way: The existing chain leaves its initial position and subsequently is attached from its bottom end to the top of the carbon adatom. The electronic, magnetic, and structural properties of these chains vertically adsorbed to h-BN depend on the number of carbon atoms in the chain, such that they exhibit an even-odd disparity. An individual carbon chain can also modify the electronic structure with localized states in the wide band gap of h-BN. As a reverse situation, we examined the growth of short BN atomic chains on graphene, which attribute diverse properties depending on whether B or N is the atom bound to the substrate. These results together with ab initio molecular dynamics simulations of the growth process reveal the interesting self-assembly behavior of the grown chains.